
Cerebrotendinous Xanthomatosis

Cerebrotendinous xanthomatosis (CTX) is a rare, autosomal, recessive neurodegenerative disorder affecting bile acid synthesis.
CAUSE AND SYMPTOMS :
CTX is associated with mutations in the cytochrome P450 CYP27A1 gene that result in production of a defective sterol 27-hydroxylase enzyme. CYP27A1 is a mitochondrial enzyme responsible for catalyzing multiple hydroxylation reactions in cholesterol metabolism and bile acid synthesis.
The impairment of CYP27A1 enzyme in CTX leads to abnormally high levels of cholestanol in the blood and the accumulation of cholestanol and cholesterol throughout the body, particularly in the brain, lens and tendons.
CTX follows a slow and progressive course, with diverse clinical presentations that may involve varied combinations of neurologic and non-neurologic manifestations with onset as early as infancy or as late as adulthood.
The main clinical manifestations are neonatal cholestatic jaundice, infantile-onset intractable, diarrhea, juvenile-onset bilateral cataracts, young adult-onset tendon xanthomas, and progressive neurologic and psychiatric dysfunction.

Chronic diarrhea

Cataracts in
both eyes
Tendon xanthomas

Neurological problems

Prevalence
The estimated prevalence of CTX is between 1:50,000 and 1:100,000 worldwide, and varies among countries and ethnic groups.

However, CTX is under-diagnosed, mainly due to its wide clinical spectrum mostly which includes non-specific manifestations and overlapping with various disorders. As a result, the actual CTX may have a higher prevalence than commonly recognized.
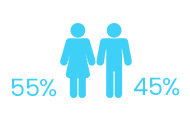
CTX has been observed more frequently among female than male individuals (~55% vs 45%).
Diagnosis :
The diagnosis of CTX relies primarily on clinical findings, biochemical testing, neuroimaging, and molecular genetic analysis. The clinical presentation can vary considerably in type, severity, and age of onset of symptoms. This variability likely contributes to undiagnosed cases or delayed diagnosis. The onset of symptoms usually occurs during childhood or adolescence, but diagnosis is often not made until adulthood. On average, patients with CTX receive a diagnosis at the age of 35, reflecting a diagnostic delay of about 16 years.Current treatment options :
The etiologic management of CTX relies principally on lifelong oral bile acid supplementation (replacement therapy) to reduce plasma cholestanol and urinary bile alcohols levels; surgery of xanthomas and other symptomatic therapies may be helpful as well.
However, advanced symptoms persisting over many years are unlikely to be significantly improved. Beginning treatment as early as possible is crucial for preventing neurological damage and deterioration in patients with CTX. Cholic acid is more rarely used instead of CDCA, especially in young children.
Depending on the manifestations, additional therapies may be necessary such as statins to treat hypercholesterolemia, antidepressant medication in case of depression, antiepileptics in case of epilepsy, levodopa in case of parkinsonism, or botulinum toxin in case of dystonia. Neonatal liver transplantation is necessary exceptionally in case of severe cholestasis.
Unmet medical needs :
Unmet medical needs in CTX remain very high:
- Clinical benefit of CDCA treatment on neuro-psychiatric outcomes is very variable and reversibility of symptoms or arrest of progression depend heavily on when replacement therapy treatment is initiated: treatment efficacy is limited when significant neurologic pathology has already manifested; moreover, up to 61% of the patients experience a decline in neurologic symptoms when treatment is initiated after the age of 24 years.
- Biologically, replacement therapy does not restore the physiological profile of bile acid intermediates.
More than 80% of patients and caregivers consider that there is a real need for new treatment for CTX.
POTENTIAL BENEFIT OF AAV GENE THERAPY
Correction through gene therapy involves the reinstatement of full CYP27A1 activity, addressing the limitations of CDCA and potentially achieving a long-lasting normalization of all parameters for a curative effect. The supplementation of CYP27A1 using an AAV vector emerges as a safe and feasible alternative for treating CTX, presenting the opportunity for comprehensive and stable metabolic correction following a single vector administration.
Publication :
– Lumbreras et al 2021 publication
Appadurai et al. Apparent underdiagnosis of cerebrotendinous xanthomatosis revealed by analysis of ~60,000 human exomes. Mol Genet Metab. 116(4) :298-304 (2015).
Nie et al. Cerebrotendinous xanthomatosis: a comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet Journal of Rare Diseases 9:179 (2014).
Salen G, Steiner RD. Epidemiology, diagnosis, and treatment of cerebrotendinous xanthomatosis (CTX). J Inherit Metab Dis. 40(6):771-781 (2017).
Stelten BML, et al. Long-term treatment effect in cerebrotendinous xanthomatosis depends on age at treatment start. Neurology. 92(2):e83-e95. doi: 10.1212/WNL.0000000000006731 (2018).
Verrips A, et al. The safety and effectiveness of chenodeoxycholic acid treatment in patients with cerebrotendinous xanthomatosis: two retrospective cohort studies. Neurol Sci. 41(4):943-949 (2019).